

The discovery of the genetic defect in cystic fibrosis three decades ago enabled the development of highly targeted therapeutics capable of correcting the function of the faulty CFTR protein, resulting in unprecedented treatment outcomes and drastic improvements in the quality of life of patients with this deadly disease.
Disease overview
Cystic fibrosis (CF) is a rare autosomal recessive disease, with an incidence of ~1 in 3,500 individuals. In the UK and US, approximately 10,000 and 30,000 people are affected by this condition, respectively. CF has a very early onset, with symptoms starting at birth or shortly after birth. While CF affects multiple organs in the body, the most common cause of morbidity and death is respiratory disease. Other complications that CF may cause include pancreatic insufficiency, kidney disease and infertility in males. At the start of the 20th century, it was rare for patients to survive beyond childhood, but significant medical advancements over the last few decades have pushed life expectancy up to the fourth decade of life.
Molecular basis of CF
Although CF was first described in 1935, it was not until 1989 when researchers identified that mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene encoding for the CFTR protein was the molecular basis of the disease. CF can be caused by multiple mutations in the CFTR gene; some of which cause a complete absence of the CFTR protein, whereas other mutations alter the structure of the CFTR protein, causing it to work less efficiently.
Healthy airways
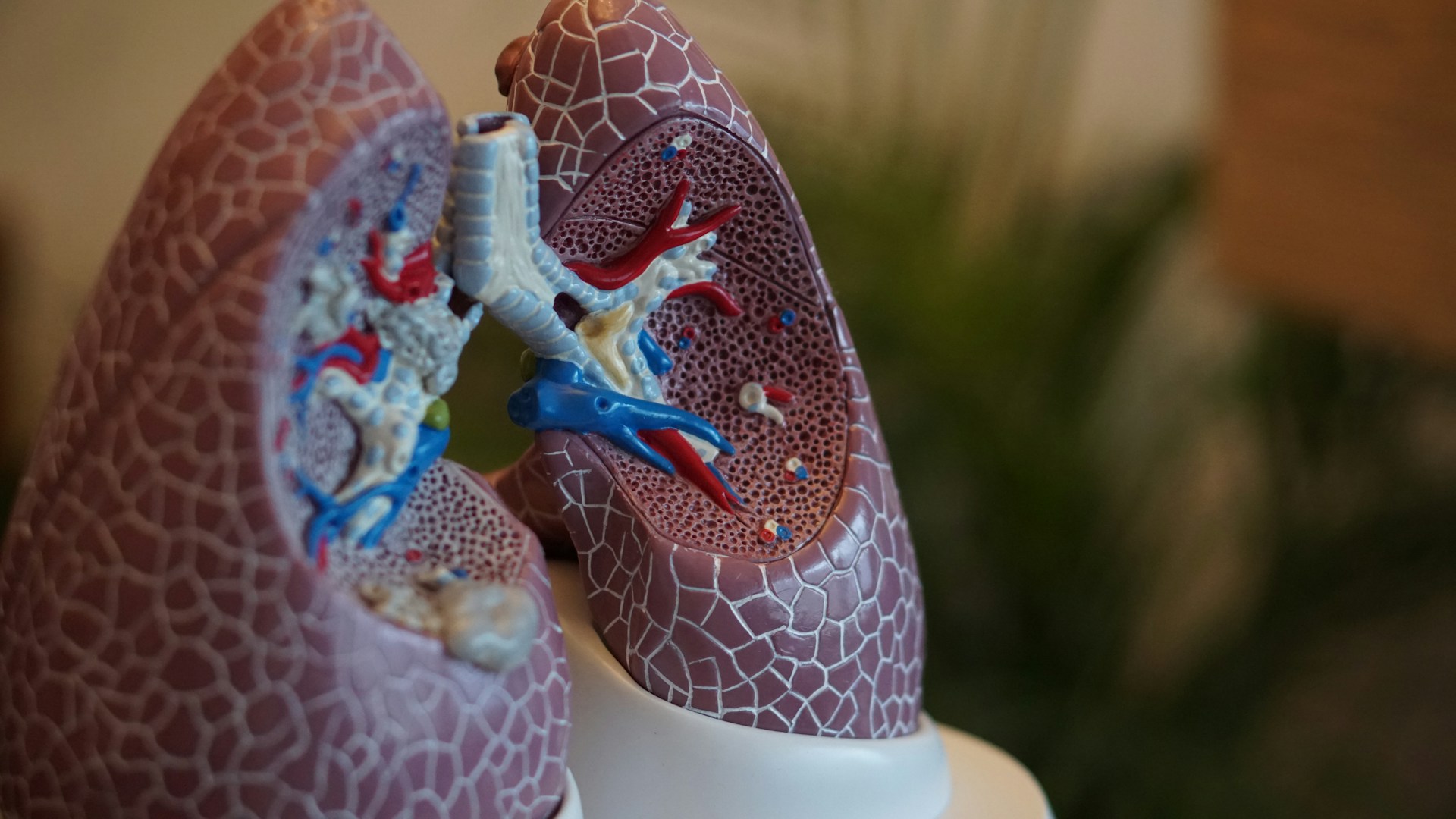
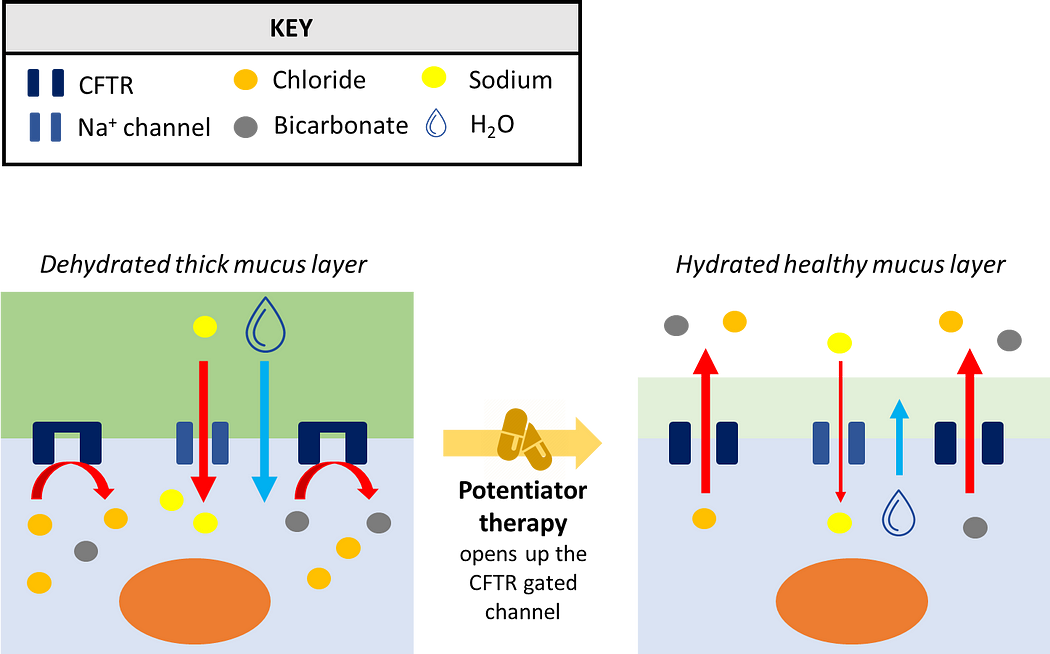
In the epithelial surface of the airways, the role of CFTR is to facilitate the flow of chloride and bicarbonate ions out of cells, which subsequently facilitates the movement of water out of cells to keep the exterior surrounding mucus layer hydrated. This keeps mucus at a desired viscosity, allowing finger-like projection known as cilia to work properly and push the mucus out of the lungs.
Airways in CF
In patients with CF, where CFTR is either missing or functioning poorly, chloride and bicarbonate ions cannot exit from the cell. Consequently, this results in a massive influx of sodium and water into the cell from outside, dehydrating the exterior mucus layer. This drastic loss of water increases the viscosity of mucus, preventing cilia from sweeping and clearing the mucus out of the lungs. As a consequence, bacteria and other microorganisms which would normally be cleared away can multiply and cause airway infection, leading to chronic inflammation and severe lung damage, which over time results in respiratory failure.
A leap in CF care
Until recently, treatment approaches in CF were focused on symptom management. However, the discovery of the genetic and protein defect in CF enabled research efforts to focus on the development of targeted and disease-modifying therapies. In recent years, promising therapies known as CFTR modulators have become available, radically transforming the care for patients with CF. The currently available CFTR modulators are subdivided into potentiators and correctors, based on how they act on the faulty CFTR protein to improve its function.
Potentiators
Potentiators are drugs that improve the function of the CFTR protein, helping to keep the gate of the channel open to facilitate and increase the flow of ions through the plasma membrane (Figure 1). Ivacaftor is an example of a potentiator and the first ever CFTR modulator to be approved. It’s used to treat patients with gating and conduction mutations in CFTR, as well as those individuals with mutations that lead to a reduced number in the CFTR protein at the plasma membrane.
Ivacaftor has been transforming the lives of patients with sustained and long-term benefits, including slower deterioration in lung function, reduced risk of lung infections and respiratory exacerbations, improvements in BMI, exercise capacity and well-being. Despite these improvements, only 5% of patients with CF have the type of mutations that ivacaftor addresses, meaning that additional targeted therapies had to be developed to address the unmet need in the wider CF population.
Correctors
Correctors are compounds indicated in patients with mutations that cause protein misfolding, which essentially cause CFTR to be sent to the proteasome where it gets destroyed. Correctors ensure adequate folding and processing of the CFTR protein, so that it can travel and be positioned at its intended location (the plasma membrane), rather than being degraded at the proteasome. Approximately 85% of CF patients have mutations that can be addressed with corrector agents.
Lumacaftor was the first corrector to be approved in combination with ivacaftor for a much wider population of patients with CF. Despite promising results in vitro, lumacaftor alone failed to show improvements in lung function. It soon became evident that the use of correctors would have to be accompanied by potentiators to maximise their efficacy. This is because while correctors ensure that the CFTR protein reaches the plasma membrane, the protein still cannot function properly. Therefore, the addition of a corrector ensures that the CFTR protein that has reached the plasma membrane stays open to enable the flow of ions. The combination of lumacaftor and ivacaftor showed significant, albeit modest, improvements in lung function.
Tezacaftor is a second generation corrector that showed improved pharmacokinetic properties and a better safety profile compared with lumacaftor. In clinical trials, tezacaftor in combination with ivacaftor showed comparable therapeutic outcomes vs lumacaftor plus ivacaftor. Despite a similar efficacy profile to lumacaftor, the improved tolerability profile and fewer drug-to-drug interactions with tezacaftor led to its approval in combination with ivacaftor.
A triple approach
As the dual combinations of lumacaftor/ivacaftor and tezacaftor/ivacaftor only showed modest efficacy outcomes, it became clear that to maximise treatment outcomes there was a need to develop novel correctors. Ultimately, the next generation corrector elexacaftor was developed and studied in combination with the backbone of tezacaftor/ivacaftor.
Instead of the previous modest improvements seen with dual combination therapies, this triple approach dramatically improved lung function by 14% and reduced pulmonary exacerbations by 63%, while being generally well tolerated. The magnitude of these benefits led to the approval of this triple therapy in the US in 2019 under the name of TRIKAFTA® and to EU approval in 2020 under the name of KAFTRIO®.
A brighter future
KAFTRIO® is a therapy that not only improves clinical outcomes, but it also drastically improves quality of life and it’s expected that patients will also live substantially longer. While the cost of CFTR modulator therapy is extremely high (€150,000 to €250,000 per year), the benefits that patients can obtain at present were simply unheard of just 10 years ago. Although the hight costs with these life-changing therapies are a concern, it is important to acknowledge that the future has never looked brighter for patients with CF.









